Pedigree Information¶
For the informatics of family data, ONETOOL utilizes PEDINFO in the S.A.G.E. package that provides many useful descriptive statistics on pedigree data, including means, standard deviations; family, sibship and pedigree sizes; and counts of each type of relative pair. This is done for ONETOOL run by default.
$ onetool --fam test_miss00.fam
$ onetool --fam test_miss00.fam --pheno test_miss0_phen.txt --pname sbp
The output file is a text file containing a formatted table with several sub-tables. Each sub-table contains the pedigree-wise/pair-wise/sibship-wise/individual-wise descriptive statistics as shown below:
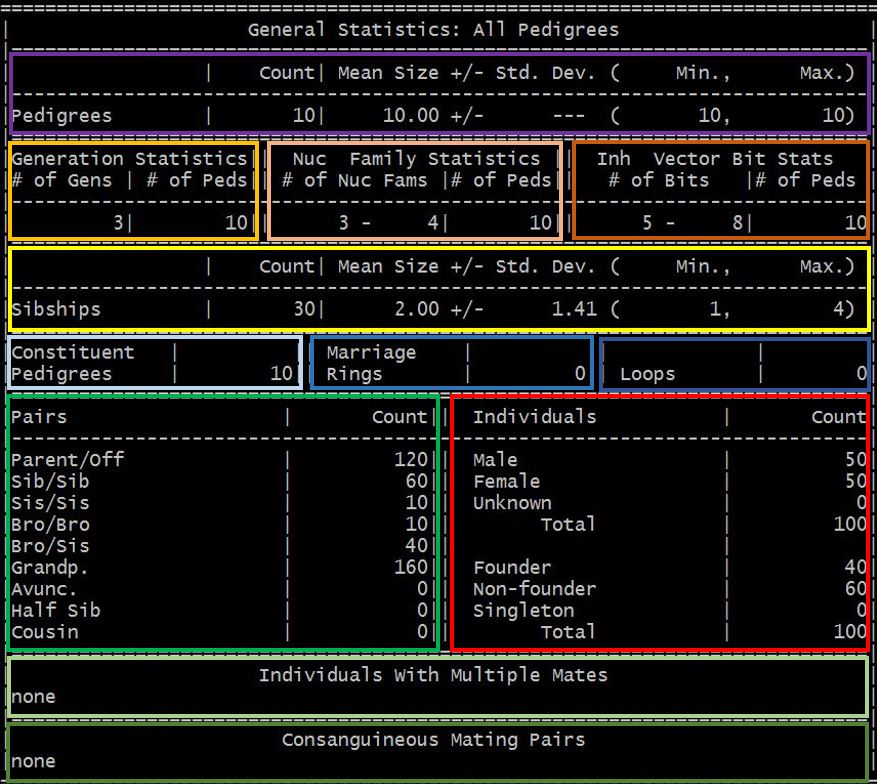
- pedigree count, mean size with standard deviation, range
- pedigree count by generation and numclear family count and inheritance vector bit size
- sibship count, mean size with standard deviation, range
- number of constituent pedigrees (i.e., disjoint sub-pedigrees), marriage rings and loops (if found)
- relative pair count by type and individual count by sex and founder/non-founder/singleton status
- individuals with multiple mates (if found)
- consanguineous mating pairs (if found)
Note
The colored-blocks are added to show the different sub-tables.
Pedigree Plot¶
ONETOOL dynamic version with R plugin provides visualization of family data utilizing the R package kinship2 to generate a plot (--plot).
$ onetool --fam test_miss00.fam --plot
Mendelian Error Check¶
Two types of errors are there in family genetic data, pedigree errors and genotyping error, which both can lead to either increased false negatives or false positives in both linkage and association studies.
Pedigree errors are the misspecified relationships among individuals in family data.
Mendelian inconsistencies can be used to identify relationship errors and genotyping error.
Mendelian inconsistencies are usually identified by comparing the genotypes of one or both parents to the genotypes of their offspring. It involves checking for each locus whether one of the two alleles that a offspring has could have been inherited from one of its parents.
ONETOOL detects Mendelian inconsistencies in pedigree data. Each marker is individually checked for inconsistencies in every pedigree. To check for Mendelian errors in given genotype data, simply add --mendel option.
$ onetool --fam test_miss00.fam --vcf test_miss00.vcf --mendel
ONETOOL reports the results by family, by sample and by variant.
- By family (.family.res)
| Column | Description |
|---|---|
| FID | Family ID |
| ERROR | Total number of alleles with Mendelian error in the family |
| AVAIL | Total number of called alleles in the family |
| PROP | Proportion of alleles with Mendelian error in the family |
- By sample (.sample.res)
| Column | Description |
|---|---|
| FID | Family ID of the sample |
| IID | Individual ID of the sample |
| ERROR | Total number of alleles with Mendelian error in the sample |
| AVAIL | Total number of called alleles in the sample |
| PROP | Proportion of alleles with Mendelian error in the sample |
- By variant (.variant.res)
| Column | Description |
|---|---|
| CHR | Chromosome name |
| VARIANT | Variant ID |
| POS | Physical position of the variant |
| ALT | An alternative allele |
| ERROR | Total number of alleles with Mendelian error |
| AVAIL | Total number of called alleles |
| PROP | Proportion of alleles with Mendelian error |
Sample Information¶
The sample-wise information from variant data helps to better understand the genetic background of the individuals in family in population level. ONETOOL calculates the following information on each sample.
- Heterozygosity
$ onetool --fam test_miss00.fam --vcf test_miss00.vcf --het
- Ratio of Heterozygote and homozygote
$ onetool --fam test_miss00.fam --vcf test_miss00.vcf --hethom
Variant Information¶
ONETOOL’s options for the variant QC and informatics are similar with those in PLINK, but they are implemented in a computationally optimized way providing more speed and efficiency.
Types of variant-wise information analysis supported:
- F-statistics - Wright’s fixation index to describe population structure
$ onetool --fam test_miss00.fam --vcf test_miss00.vcf --pheno test_miss00.pheno --pname ethnicity --fst
- Allele frequency - Minor allele frequency
$ onetool --fam test_miss00.fam --vcf test_miss00.vcf --freq
- HWE - Hardy-Weinberg Equlibrium
$ onetool --fam test_miss00.fam --vcf test_miss00.vcf --hwe
- PCA - Principle component analysis
$ onetool --fam test_miss00.fam --vcf test_miss00.vcf --pca --npc 5Note
To specify the number of principal components to compute, use --npc.